强化生物安全国家药监局修订血液制品GMP附录
属于国家管控危险化学品,根据相关法规和网安部门规定,本网站不提供该产品相关销售信息。
2019年底发生的新冠病毒肺炎疫情,对于中国民众生活的巨大影响和对于中国经济的巨大影响是毋庸讳言的。不仅如此,随着国际交往的频繁发生以及国外政府对于疫情的应对失措,新冠疫情开始在境外多国发生。随着中国境内的疫情被基本控制,国内从政府到民众都开始反思本次疫情的经验和教训。通过这次疫情,中国的最高领导层意识到必须在国家层面加强生物安全制度建设、持续加强生物安全防控力度的必要性。
2020年2月14日,据新华社报道,中央全面深化改革委员会第十二次会议强调:完善重大疫情防控体制机制健全国家公共卫生应急管理体系。在这篇新闻中,提到重要信息:国家领导人强调,要强化公共卫生法治保障,全面加强和完善公共卫生领域相关法律和法规建设,认真评估传染病防治法、野生动物保护法等法律和法规的修改完善。要从保护人民健康、保障国家安全、维护国家长治久安的高度,把生物安全纳入国家安全体系,系统规划国家生物安全风险防控和治理体系建设,全方面提高国家生物安全治理能力。要尽快推动出台生物安全法,加快构建国家生物安全法律和法规体系、制度保障体系。
随之,2020年2月15日新闻中又提到:科技部社会持续健康发展科技司司长吴远彬15日下午在国务院联防联控机制发布会上介绍,科技部出台《关于加强新冠病毒高等级病毒微生物实验室生物安全管理的指导意见》,要求实验室发挥平台作用,服务科技攻关需求,各主管部门要加强对实验室,特别是对病毒的管理,确保生物安全。
通过上面这一些信息,我们该理解到,关于本次疫情的整理和反思,以及以后要改善提高的工作,中央已经做出了部署。而作为全国药品监管工作的主体部门,国家药监局不仅在疫情期间积极审批器械和药品,为抗疫做贡献;而且也开始启动相关法规和指南的修订工作。例如2020版药典中涉及生物安全的内容开始增加。
2020年4月15日,国家药监局下属审核查验中心(CFDI)发布了《食品药品审核查验中心公开征求《药品生产质量管理规范血液制品附录(征求意见稿)》意见的公告》,对这份管理血液制品生产管理和质量管理的重要法规开始修订。
目前血液制品行业执行的GMP附录是在2011年3月1日发布的《药品生产质量管理规范》(2010年修订)的附件4血液制品;这份文件距今将近10年时间了。可以说,已经在多个角度不能够满足目前的血液制品监管需要了。
谈到生物安全,应该说在2019年初发生的上海新兴事件,已经为制药行业敲响了警钟。在2019年2月6日发布的《国家药监局公布上海新兴相关这类的产品初步调查情况》提到:2月5日,国家药监局接到国家卫生健康委通报,上海新兴医药股份有限公司(以下简称上海新兴)生产的一批次静注人免疫球蛋白(批号:20180610Z)艾滋病病毒抗体检测为阳性。国家药监局立即要求上海市药监局对上海新兴开展现场检查,组织对同批原料血浆生产的产品和相邻批次产品做检验,并派出督导检查组抵达上海、江西,督促指导地方药监部门开展调查处置工作。国家药监局、国家卫生健康委联合下发通知,要求立即暂停使用该公司制作的相关这类的产品,并及时采取封存等控制措施。上海方面对涉事批次静注人免疫球蛋白进行的艾滋病、乙肝、丙肝三种病毒核酸检验测试,结果均为阴性;江西方面对患者的艾滋病病毒核酸检验测试,结果为阴性。对于上海新兴事件的反思和本次新冠疫情的反思,都促使国家药监局加快相关的修订。
例如《血液制品》征求意见稿的第四条提到:血液制品的管理还应当符合本规范其他适用附录和国家相关规定。在原文件上面只是提到要符合国家相关规定,没有涉及和GMP配套的其他附录。事实上,如果要实施《血液制品》GMP附录,是不可能脱离其他GMP附录的。例如洁净区设置需要参考附录1《无菌药品》,产品取样需要参考《取样》附录,各项验证工作需要参考《确认和验证》附录,计算机化系统管理需要参考《计算计化系统》。
例如《血液制品》征求意见稿继续和现行版文件保持一致,对公司负责人提出合规高要求。例如第六条提到:企业负责人应当具有血液制品专业相关知识,并经过有关规定法律知识的培训。从法规角度要求血液制品的负责人一定要重视法规和法律学习,强化合规责任。
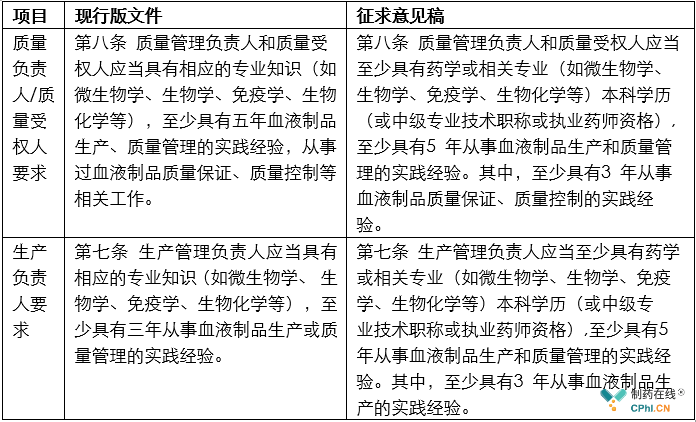
从上面表格能看出,新的征求意见稿对于血液制品行业的关键从业人员的要求,不管是职称还是从业年限,都进行了提高。
征求意见稿对于血液制品公司的生产设施和检验设施都提出更加高的要求。例如下面这些条款都在原来要求的基础上,提出了更加高的要求,强化数据规范,各类设施充分隔离,避免交叉污染。
第十二条:原料血浆、血液制品检验实验室应当符合国务院《病原微生物实验室生物安全管理条例》、国家标准《实验室生物安全通用要求》的有关法律法规,并具备与公司制作和质量发展要求相适应的检验能力。企业应定期开展实验室能力评估,确保实验结果准确、可靠和检验过程信息记录的真实、准确、完整和可追溯
第十三条:原料血浆、尚未经过病毒去除和/或灭活处理的中间产品检验实验室应当独立设置,使用专用检验设备,并应当有原位灭活或消毒的设备。如有空调系统,应当独立设置。
第十四条:原料血浆破袋、合并、分离、提取、分装前的巴氏灭活等工序至少在D 级洁净区内进行。

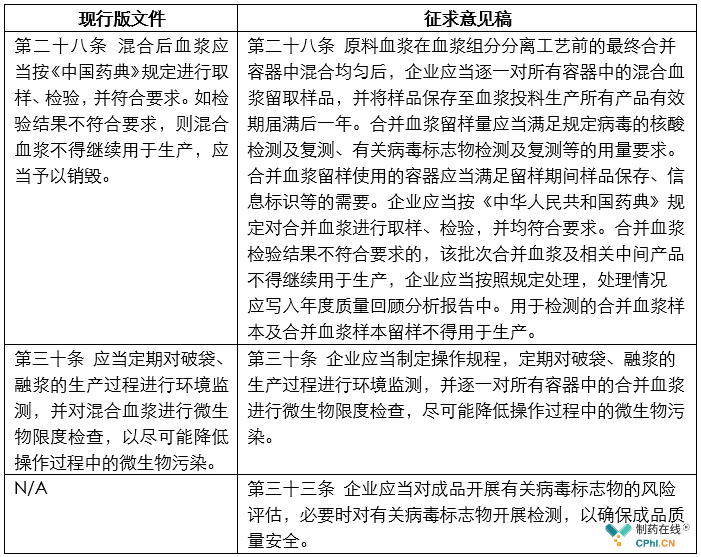
从上面对比表格内容看,不仅在血浆入厂检验、中控检验和日常监管措施中进行了强化管理,新的征求意见稿还增加一个新的33条:应当对成品开展有关病毒标志物的风险评估,必要时对有关病毒标志物开展检测,以确保成品质量安全。
从上面汇总分析的内容看,这次修订的《血液制品》GMP附录征求意见稿在人员、组织、供应商监管、关键物料入厂控制、厂房设施隔离等角度,进行全面发力,以达到确保血液制品质量稳定,并对从业人员进行保护的目的。
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势变化分析和制药企业并购项目的风险管理工作。

质量管理体系GMP指南问答第3期(上) 谁是质量管理体系有效运行的关键?
更多
本周,有不少热点需要我们来关注。审评审批方面,多个药物获批。最值...[详细]
2023年12月22日,中国食品药品检定研究院(以下简称中检院)官网...[详细]
全球首 款PD-1/CTLA-4双抗申报新适应症,一线日,康方生物「卡度尼利单抗注射液」的2.2类注册申请获CDE受...[详细]
过去的一年,据NMPA官网批件信息统计,共有80余款新药在国内首次...[详细]
12月11日,FDA公布了一则召回通告。InvaGen Pharmaceuticals的一款氨己烯...[详细]